COORDINACION
Coordinación
Se entiende por número de coordinación el número de vecinos inmediatos de un átomo, y por poliedro de coordinación, el poliedro resultante de la unión de los centros de los átomos de coordinación. Así, si un átomo está rodeado de tres átomos vecinos formando un triangulo plano, el número de coordinación és tres, y el poliedro (en este caso plano) un triangulo.
Se entiende por número de coordinación el número de vecinos inmediatos de un átomo, y por poliedro de coordinación, el poliedro resultante de la unión de los centros de los átomos de coordinación. Así, si un átomo está rodeado de tres átomos vecinos formando un triangulo plano, el número de coordinación és tres, y el poliedro (en este caso plano) un triangulo.

La coordinación en una estructura en que las partículas estructurales (las que se enlazan para formar la estructura cristalina) son átomos, depende de si el enlace es direccional o no.

En el caso de los enlaces direccionales (covalente), el número de vecinos de un átomo depende del número de orbitales de enlace, y el poliedro de coordinación, de las direcciones de estos orbitales. Por el contrario, en los casos de los enlaces no direccionales (metálico e iónico), como que la estructura más estable es la más compacta, el número y el poliedro de coordinación depende de los tamaños relativos de los átomos de la estructura: cada catión se rodea de tantos aniones como sea posible en función de sus tamaños relativos.
A continuación se hace un análisis de los posibles casos de coordinación en los enlaces NO direccionales:
a) si la diferencia de tamaños es muy grande, alrededor de un catión no hay lugar para tres aniones, y por tanto, solo habrá dos. En este caso la coordinación es lineal, y el número de coordinación dos.
En la figura se muestra este caso en una diagrama esquemático:
La estructura estable es de coordinación lineal porqué si fuese una coordinación mayor, por ejemplo 3, el átomo central "bailaría" en el espacio intersticial entre los aniones y la entropía de la disposición estructural a lo mejor sería demasiado grande.
b) para que el catión tenga coordinación 3, debe ser tangente simultáneamente a los tres aniones, como se muestra en la siguiente figura
a) si la diferencia de tamaños es muy grande, alrededor de un catión no hay lugar para tres aniones, y por tanto, solo habrá dos. En este caso la coordinación es lineal, y el número de coordinación dos.
En la figura se muestra este caso en una diagrama esquemático:
La estructura estable es de coordinación lineal porqué si fuese una coordinación mayor, por ejemplo 3, el átomo central "bailaría" en el espacio intersticial entre los aniones y la entropía de la disposición estructural a lo mejor sería demasiado grande.
b) para que el catión tenga coordinación 3, debe ser tangente simultáneamente a los tres aniones, como se muestra en la siguiente figura


En el esquema de la derecha se puede comprobar que se cumple:

donde R y r son los radios del anión y del catión, respectivamente.
De aquí se deduce que r = 0.155R .
Esta relación es el límite inferior para que tenga lugar la coordinación triangular. Por debajo de este tamaño relativo habrá coordinación lineal, y por encima triangular, por bien que con tamaños ligeramente superiores, los tres
aniones no serán tangentes entre ellos, y solo lo serán con el catión.
De aquí se deduce que r = 0.155R .
Esta relación es el límite inferior para que tenga lugar la coordinación triangular. Por debajo de este tamaño relativo habrá coordinación lineal, y por encima triangular, por bien que con tamaños ligeramente superiores, los tres
aniones no serán tangentes entre ellos, y solo lo serán con el catión.

c) Si el catión es mayor, puede llegar a un tamaño tal que quepan cuatro aniones a su alrededor, y por tanto se alcanza la coordinación cuatro. En estas
circunstancias, la disposición que tendría lugar es aquella que mantenga los aniones lo más separados posible para que se minimice la repulsión electrostática entre ellos: esta forma es un tetraedro, de manera que los centros de los aniones ocupan el vértice de un tetraedro, en el centro del cual está el catión.
circunstancias, la disposición que tendría lugar es aquella que mantenga los aniones lo más separados posible para que se minimice la repulsión electrostática entre ellos: esta forma es un tetraedro, de manera que los centros de los aniones ocupan el vértice de un tetraedro, en el centro del cual está el catión.

Si se hace una sección para un de los planos diagonales del cubo de la figura de la izquierda, se obtiene el esquema de la derecha, en el cual




de donde se deduce que , que representa el límite inferior para r = 0.225R la coordinación tetraédrica, y por tanto se puede afirmar que entre las relaciones 0.155R y 0.225 R existirá coordinación triangular, mientras que por encima de 0.225R la coordinación será tetraédrica.
d) Para cationes de tamaños relativos respecto de los aniones más grandes puede llegar a darse coordinación octaédrica, es decir seis aniones alrededor del catión, los centros de los cuales dibujan un octaedro de coordinación alrededor del catión central. Haciendo una sección que pase por el centro de este octaedro queda un esquema como el de la figura siguiente:
d) Para cationes de tamaños relativos respecto de los aniones más grandes puede llegar a darse coordinación octaédrica, es decir seis aniones alrededor del catión, los centros de los cuales dibujan un octaedro de coordinación alrededor del catión central. Haciendo una sección que pase por el centro de este octaedro queda un esquema como el de la figura siguiente:
En la cual:

donde a es la resta del octaedro, y de donde se deduce r = 0,414R que es el límite inferior para la coordinación octaédrica, de manera que entre esta relación y la anteriormente determinada habrá coordinación tetraédrica.
e) La siguiente coordinación posible es cúbica, con número de coordinación 8, como se muestra en la figura, una sección diagonal la cual permite establecer la relación de tamaños de radios necesaria para que se dé esta coordinación En la figura de la derecha se puede deducir:
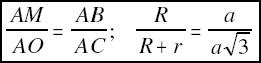
e) La siguiente coordinación posible es cúbica, con número de coordinación 8, como se muestra en la figura, una sección diagonal la cual permite establecer la relación de tamaños de radios necesaria para que se dé esta coordinación En la figura de la derecha se puede deducir:

y de aquí r = 0.732R que, como en los otros casos, es el límite inferior para la
coordinación cúbica.
f) Si los tamaños de los átomos son iguales, la coordinación que se da es 12, con un poliedro de coordinación, la forma del cual se puede ver en la siguiente figura, en que los átomos que rodean el central ocupan las posiciones de los centros de las aristas de un cubo:
Y considerando uno de los triángulos en que se divide el hexágono dibujado en la figura con líneas marrones:
coordinación cúbica.
f) Si los tamaños de los átomos son iguales, la coordinación que se da es 12, con un poliedro de coordinación, la forma del cual se puede ver en la siguiente figura, en que los átomos que rodean el central ocupan las posiciones de los centros de las aristas de un cubo:
Y considerando uno de los triángulos en que se divide el hexágono dibujado en la figura con líneas marrones:




Y como que sin30º=0.5, hay que concluir que r=R

Es decir que para que se de coordinación 12 hace falta que todas las esferas a empaquetar sean de la misma medida.










 +
+ →
→ +
+ +
+






{kind=link}